The field of bioinformatics applies computational techniques to biology. This study focuses in particular on proteins, which are large molecules that have specific functions in organisms. Understanding proteins requires identifying fixed patterns called motifs in protein sequences, as motifs are indicative of a protein’s structure and function.
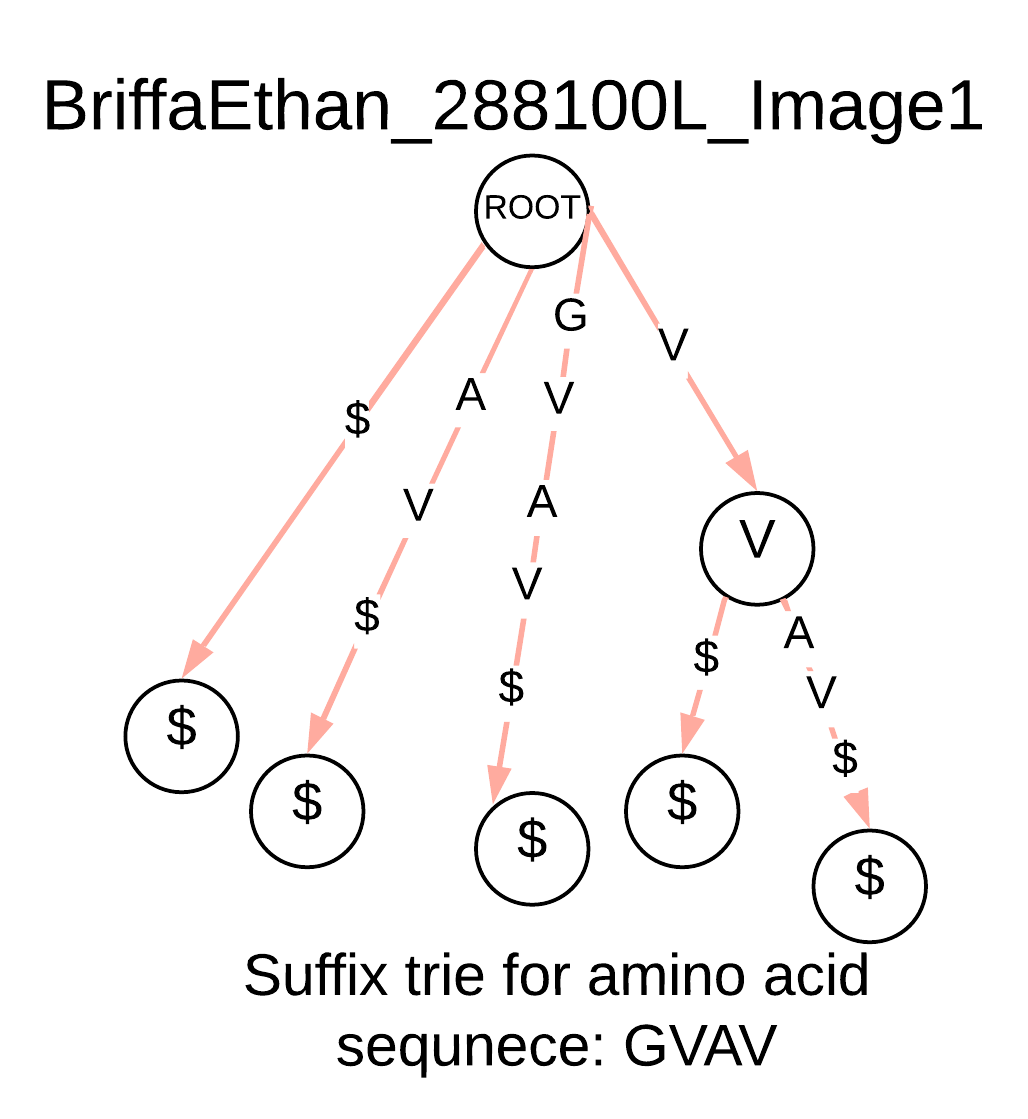
This research attempts to improve the speed of finding motifs by comparing unknown protein sequences with known protein domains as classified in the CATH hierarchy. The approach adopted in this study uses the multiple sequence alignment (MSA) from proteins found in CATH functional families. Each MSA contains motifs having sequence regions that have been preserved through evolution, known as conserved regions. The representative sequences for the functional families are stored as a suffix trie, which would then be used to find potential structures.
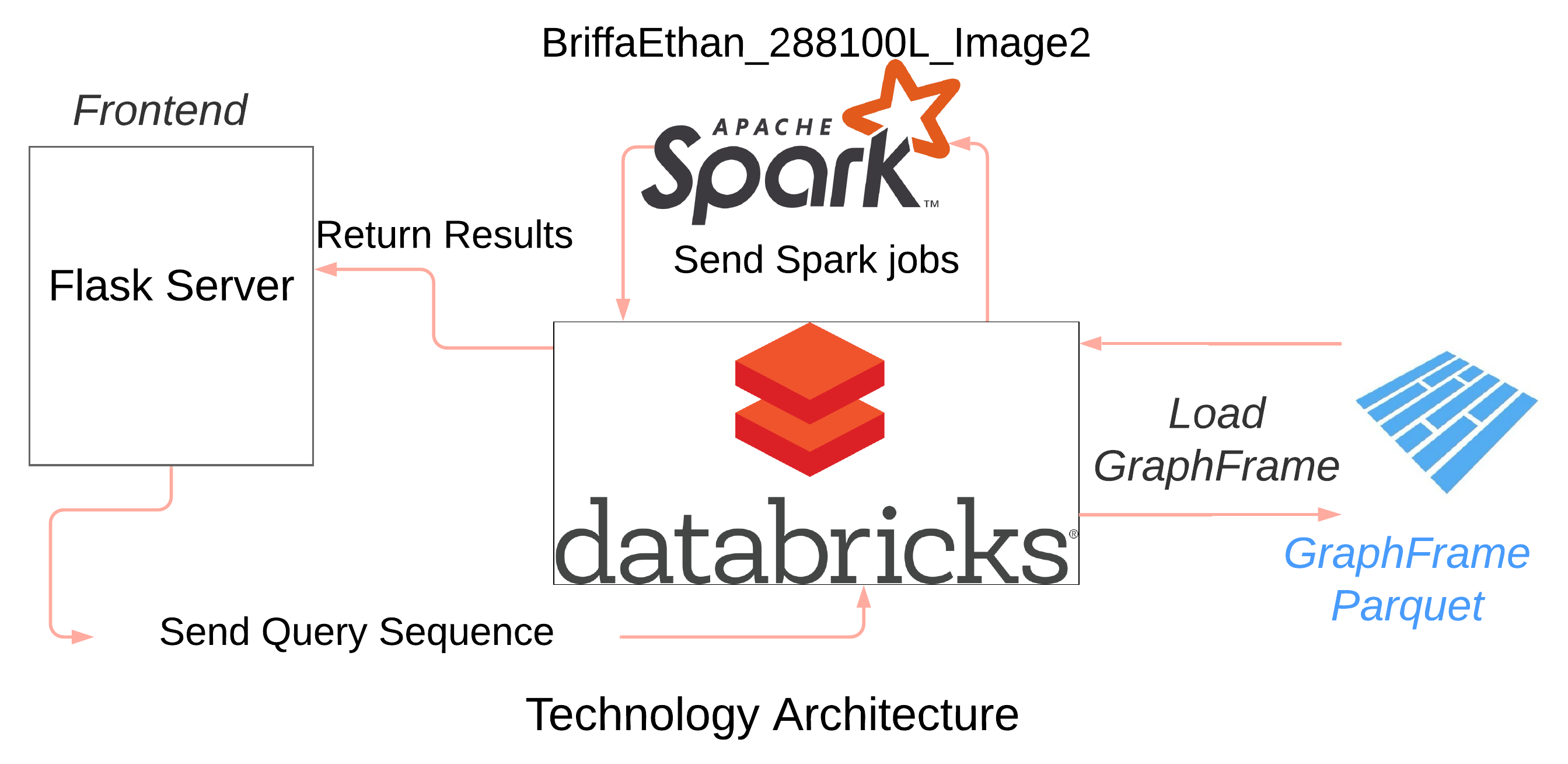
To improve the efficiency of the search, the suffix trie is implemented using the Apache Spark framework, which is generally used to process large amounts of data efficiently. The Spark architecture offers processing scalability by distributing the process over a number of nodes, thereby speeding up the search.
The method subsequently determines the best match through a scoring algorithm, which ranks the output based on the closest match to a known structural motif. A substitution matrix is also used to consider all possible variations of the conserved regions.
This system was finally compared against a library of hidden Markov models, where the results were compared to determine the speed of its process whilst ensuring that the correct results would be produced.
Course: B.Sc. IT (Hons.) Software Development
Supervisor: Mr Joseph Bonello